Metagenome Assembled Genomes Workflow (v1.0.2)
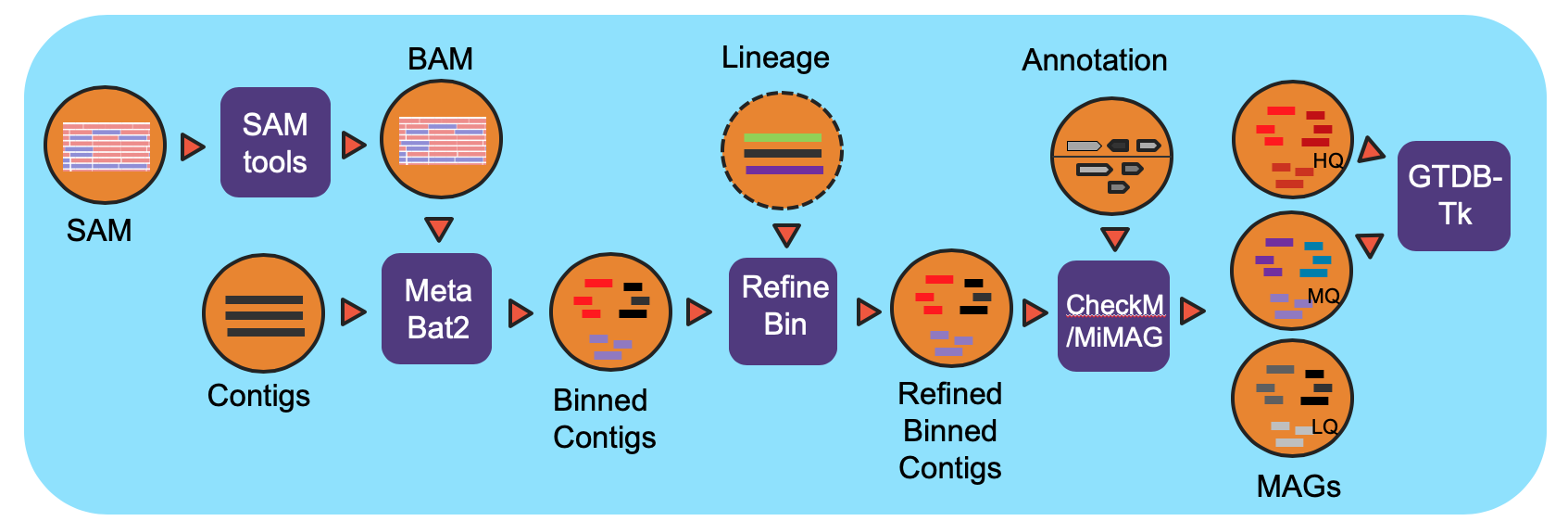
Workflow Overview
The workflow is based on IMG metagenome binning pipeline and has been modified specifically for the NMDC project. For all processed metagenomes, it classifies contigs into bins using MetaBat2. Next, the bins are refined using the functional Annotation file (GFF) from the Metagenome Annotation workflow and optional contig lineage information. The completeness of and the contamination present in the bins are evaluated by CheckM and bins are assigned a quality level (High Quality (HQ), Medium Quality (MQ), Low Quality (LQ)) based on MiMAG standards. In the end, GTDB-Tk is used to assign lineage for HQ and MQ bins.
Workflow Availability
The workflow from GitHub uses all the listed docker images to run all third-party tools. The workflow is available in GitHub: https://github.com/microbiomedata/metaMAGs The corresponding Docker image is available in DockerHub: https://hub.docker.com/r/microbiomedata/nmdc_mbin
Requirements for Execution
(recommendations are in bold):
WDL-capable Workflow Execution Tool (Cromwell)
Container Runtime that can load Docker images (Docker v2.1.0.3 or higher)
Hardware Requirements
Disk space: > 27 GB for the CheckM and GTDB-Tk databases
Memory: ~120GB memory for GTDB-tk.
Workflow Dependencies
Third party software (These are included in the Docker image.)
Biopython v1.74 (License: BSD-3-Clause)
Sqlite (License: Public Domain)
Pymysql (License: MIT License)
requests (License: Apache 2.0)
samtools > v1.9 (License: MIT License)
Metabat2 v2.15 (License: BSD-3-Clause)
CheckM v1.1.2 (License: GPLv3)
GTDB-TK v1.2.0 (License: GPLv3)
FastANI v1.3 (License: Apache 2.0)
FastTree v2.1.10 (License: GPLv2)
Requisite databases
The GTDB-Tk database must be downloaded and installed. The CheckM database included in the Docker image is a 275MB file contains the databases used for the Metagenome Binned contig quality assessment. The GTDB-Tk (27GB) database is used to assign lineages to the binned contigs.
The following commands will download and unarchive the GTDB-Tk database:
wget https://data.ace.uq.edu.au/public/gtdb/data/releases/release89/89.0/gtdbtk_r89_data.tar.gz tar -xvzf gtdbtk_r89_data.tar.gz mv release89 GTDBTK_DB rm gtdbtk_r89_data.tar.gz
Sample dataset(s)
The following test dataset include an assembled contigs file, a BAM file, and a functional annotation file: metaMAGs_test_dataset.tgz
Input
A JSON file containing the following:
the number of CPUs requested
The number of threads used by pplacer (Use lower number to reduce the memory usage)
the path to the output directory
the project name
the path to the Metagenome Assembled Contig fasta file (FNA)
the path to the Sam/Bam file from read mapping back to contigs (SAM.gz or BAM)
the path to contigs functional annotation result (GFF)
the path to the text file which contains mapping of headers between SAM or BAM and GFF (ID in SAM/FNA<tab>ID in GFF)
the path to the database directory which includes checkM_DB and GTDBTK_DB subdirectories.
(optional) scratch_dir: use –scratch_dir for gtdbtk disk swap to reduce memory usage but longer runtime
An example JSON file is shown below:
{
"nmdc_mags.cpu":32,
"nmdc_mags.pplacer_cpu":1,
"nmdc_mags.outdir":"/path/to/output",
"nmdc_mags.proj_name":" Ga0482263",
"nmdc_mags.contig_file":"/path/to/Ga0482263_contigs.fna ",
"nmdc_mags.sam_file":"/path/to/pairedMapped_sorted.bam ",
"nmdc_mags.gff_file":"/path/to/Ga0482263_functional_annotation.gff",
"nmdc_mags.map_file":"/path/to/Ga0482263_contig_names_mapping.tsv",
"nmdc_mags.gtdbtk_database":"/path/to/GTDBTK_DB"
"nmdc_mags.scratch_dir":"/path/to/scratch_dir"
}
Output
The workflow creates several output directories with many files. The main output files, the binned contig files from HQ and MQ bins, are in the hqmq-metabat-bins directory; the corresponding lineage results for the HQ and MQ bins are in the gtdbtk_output directory.
A partial JSON output file is shown below:
|-- MAGs_stats.json
|-- 3300037552.bam.sorted
|-- 3300037552.depth
|-- 3300037552.depth.mapped
|-- bins.lowDepth.fa
|-- bins.tooShort.fa
|-- bins.unbinned.fa
|-- checkm-out
| |-- bins/
| |-- checkm.log
| |-- lineage.ms
| `-- storage
|-- checkm_qa.out
|-- gtdbtk_output
| |-- align/
| |-- classify/
| |-- identify/
| |-- gtdbtk.ar122.classify.tree -> classify/gtdbtk.ar122.classify.tree
| |-- gtdbtk.ar122.markers_summary.tsv -> identify/gtdbtk.ar122.markers_summary.tsv
| |-- gtdbtk.ar122.summary.tsv -> classify/gtdbtk.ar122.summary.tsv
| |-- gtdbtk.bac120.classify.tree -> classify/gtdbtk.bac120.classify.tree
| |-- gtdbtk.bac120.markers_summary.tsv -> identify/gtdbtk.bac120.markers_summary.tsv
| |-- gtdbtk.bac120.summary.tsv -> classify/gtdbtk.bac120.summary.tsv
| `-- ..etc
|-- hqmq-metabat-bins
| |-- bins.11.fa
| |-- bins.13.fa
| `-- ... etc
|-- mbin-2020-05-24.sqlite
|-- mbin-nmdc.20200524.log
|-- metabat-bins
| |-- bins.1.fa
| |-- bins.10.fa
| `-- ... etc
Below is an example of all the output directory files with descriptions to the right.
FileName/DirectoryName |
Description |
|---|---|
1781_86104.bam.sorted |
sorted input bam file |
1781_86104.depth |
the contig depth coverage |
1781_86104.depth.mapped |
the name mapped contig depth coverage |
MAGs_stats.json |
MAGs statistics in json format |
bins.lowDepth.fa |
lowDepth (mean cov <1 ) filtered contigs fasta file by metaBat2 |
bins.tooShort.fa |
tooShort (< 3kb) filtered contigs fasta file by metaBat2 |
bins.unbinned.fa |
unbinned fasta file |
metabat-bins/ |
initial metabat2 binning result fasta output directory |
checkm-out/bins/ |
hmm and marker genes analysis result directory for each bin |
checkm-out/checkm.log |
checkm run log file |
checkm-out/lineage.ms |
lists the markers used to assign taxonomy and the taxonomic level to which the bin |
checkm-out/storage/ |
intermediate file directory |
checkm_qa.out |
checkm statistics report |
hqmq-metabat-bins/ |
HQ and MQ bins contigs fasta files directory |
gtdbtk_output/identify/ |
gtdbtk marker genes identify result directory |
gtdbtk_output/align/ |
gtdbtk genomes alignment result directory |
gtdbtk_output/classify/ |
gtdbtk genomes classification result directory |
gtdbtk_output/gtdbtk.ar122.classify.tree |
archaeal reference tree in Newick format containing analyzed genomes (bins) |
gtdbtk_output/gtdbtk.ar122.markers_summary.tsv |
summary tsv file for gtdbtk marker genes identify from the archaeal 122 marker set |
gtdbtk_output/gtdbtk.ar122.summary.tsv |
summary tsv file for gtdbtk archaeal genomes (bins) classification |
gtdbtk_output/gtdbtk.bac120.classify.tree |
bacterial reference tree in Newick format containing analyzed genomes (bins) |
gtdbtk_output/gtdbtk.bac120.markers_summary.tsv |
summary tsv file for gtdbtk marker genes identify from the bacterial 120 marker set |
gtdbtk_output/gtdbtk.bac120.summary.tsv |
summary tsv file for gtdbtk bacterial genomes (bins) classification |
gtdbtk_output/gtdbtk.bac120.filtered.tsv |
a list of genomes with an insufficient number of amino acids in MSA |
gtdbtk_output/gtdbtk.bac120.msa.fasta |
the MSA of the user genomes (bins) and the GTDB genomes |
gtdbtk_output/gtdbtk.bac120.user_msa.fasta |
the MSA of the user genomes (bins) only |
gtdbtk_output/gtdbtk.translation_table_summary.tsv |
the translation table determined for each sgenome (bins) |
gtdbtk_output/gtdbtk.warnings.log |
gtdbtk warning message log |
mbin-2021-01-31.sqlite |
sqlite db file stores MAGs metadata and statistics |
mbin-nmdc.20210131.log |
the mbin-nmdc pipeline run log file |
rc |
cromwell script sbumit return code |
script |
Task run commands |
script.background |
Bash script to run script.submit |
script.submit |
cromwell submit commands |
stderr |
standard error where task writes error message to |
stderr.background |
standard error where bash script writes error message to |
stdout |
standard output where task writes error message to |
stdout.background |
standard output where bash script writes error message to |
complete.mbin |
the dummy file to indicate the finish of the pipeline |
Version History
1.0.2 (release date 02/24/2021; previous versions: 1.0.1)
Point of contact
Original author: Neha Varghese <njvarghese@lbl.gov>
Package maintainer: Chienchi Lo <chienchi@lanl.gov>