Metabolomics Workflow (v2.1.0)
Summary
The gas chromatography-mass spectrometry (GC-MS) based metabolomics workflow (metaMS) has been developed by leveraging PNNL’s CoreMS software framework. The current software design allows for the orchestration of the metabolite characterization pipeline, i.e., signal noise reduction, m/z based Chromatogram Peak Deconvolution, abundance threshold calculation, peak picking, spectral similarity calculation and molecular search, similarity score calculation, and confidence filtering, all in a single step.
Workflow Diagram
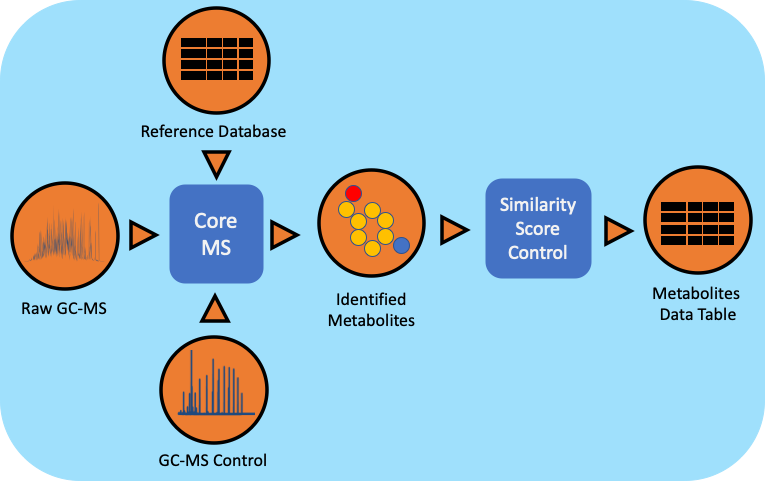
Workflow Dependencies
Third party software
CoreMS (2-clause BSD)
Click (BSD 3-Clause “New” or “Revised” License)
Database
PNNL GC-MS Spectral Database
Workflow Availability
The workflow is available in GitHub: https://github.com/microbiomedata/metaMS
The container is available at Docker Hub (microbiomedata/metaMS): https://hub.docker.com/r/microbiomedata/metams
The python package is available on PyPi: https://pypi.org/project/metaMS/
The databases are available by request. Please contact NMDC (support@microbiomedata.org) for access.
Test datasets
https://github.com/microbiomedata/metaMS/blob/master/data/GCMS_FAMES_01_GCMS-01_20191023.cdf
Execution Details
Please refer to:
https://github.com/microbiomedata/metaMS/blob/master/README.md#usage
Inputs
ANDI NetCDF for GC-MS (.cdf)
CoreMS Hierarchical Data Format (.hdf5)
CoreMS Parameter File (.JSON)
MetaMS Parameter File (.JSON)
Outputs
- Metabolites data-table
CSV, TAB-SEPARATED TXT
HDF: CoreMS HDF5 format
XLSX : Microsoft Excel
- Workflow Metadata:
JSON
Requirements for Execution
Docker Container Runtime
Python Environment >= 3.6
Python Dependencies are listed on requirements.txt
Version History
2.1.0
Point of contact
Package maintainer: Yuri E. Corilo <corilo@pnnl.gov>